Genereller Ansatz
Wer kennt das nicht?
Plötzlich sind die chromatographischen Ergebnisse verändert. Beispielsweise treten Peak-Splitting oder -Tailing auf. Vielleicht sind die Retentionszeiten verschoben oder auch gar keine Peaks detektiert.
Dann ist es an der Zeit, die Ursache der Anomalie zu finden und zu beheben. Da das endgültige Chromatogramm von verschiedenen Faktoren beeinflusst wird, sollten alle möglichen Komponenten und Variablen systematisch untersucht werden.
Eine methodische und zielgerichtete Überprüfung der folgenden Parameter ist angebracht; Jedes Verfahren wird später in diesem Dokument ausführlicher beschrieben:

Hier geht es zu Ihrem YMC (U)HPLC Troubleshooting Guide:

Probleme und deren Ursache
Rückdruckveränderung
Die Ursachen für eine Änderungen im Rückdruck sind häufig auf einen fehlerhaften Umgang mit den Proben und Lösungsmitteln zurückzuführen, der letztendlich in einer Blockierung oder Kontamination von Systemkomponenten und Säule resultiert.

Kein Druck / zu niedriger Druck
| Mögliche Ursachen | Lösungen |
| Unterdruckbildung im Eluentenvorratsgefäß |
|
| Undichtigkeiten an Kolbendichtungen oder Ventilen der Pumpe |
|
| Luft oder Partikel im Pumpenkopf oder Ventilen der Pumpe |
|
| Leakage in Übertragungsleitungen |
|
Druckanstieg / zu hoher Druck
| Mögliche Ursachen | Lösungen |
| Verstopfung von Injektor oder Kapillarwegen |
|
| Verunreinigung von Wasser durch Algen / Bakterien |
|
| blockierte Vorsäule oder Einlassfritte der Säule |
|
| Kontamination der stationären Phase |
|
Pressure Fluctuation
| Mögliche Ursachen | Lösung |
| Undichtigkeiten an Kolbendichtungenoder Ventilen der Pumpe |
|
| Luft oder Partikel im Pumpenkopf oder Ventilen der Pumpe |
|

Peakverbreiterung
| Mögliche Ursachen | Lösung |
| Zu großes Injektionsvolumen |
|
| Detektionsrate zu niedrig |
|
| Zu lange Retentionszeiten |
|
| Zu hohe Viskosität des Laufmittels |
|
| Kontamination der stationären Phase |
|
Fronting
| Mögliche Ursachen | Lösung |
| Überladung der Säule |
|
| Zu hohe Viskosität der Probe oder mobilen Phase |
|
| Kontamination der stationären Phase |
|
Tailing
| Mögliche Ursachen | Lösung |
| Wechselwirkungen mit Silanolgruppen bei basischen Analyten |
|
| Totvolumen |
|
| Alterung des Packmaterials durch zu hohe Temperaturen |
|
| Blockierte Vorsäule oder Einlassfritte der Säule |
|
| Falscher pH-Wert der mobilen Phase |
|
| Koeluierender Analyt |
|
| Kontamination der stationären Phase |
|
Doppelpeaks
| Mögliche Ursachen | Lösung |
| Blockierte Vorsäule oder Einlassfritte der Säule |
|
| Überladung der Säule |
|
| Ungeeignetes Injektions-Lösungsmittel |
|
| Totvolumen |
|
| Elution einer zweiten Probenkomponente |
|
| Carry-Over der letzten Analyse |
|
Retentionszeit und Auflösung
Eine Veränderung der Retentionszeiten und Auflösung geht häufig mit einer Änderung der Peaksymmetrie und Effizienz einher. Diese Symptome gehören zu einer normalen Alterung
der Trennsäule, können aber auch durch Leckagen im System, Probleme mit dem Injektor, instabiler Temperatur und Kontamination oder Beschädigung der stationären Phase durch ungeeignete Methodenparameter hervorgerufen werden.

Retentionszeit ist verkürzt
| Mögliche Ursachen | Lösung |
| Überladung der Säule (einhergehend mit zu hohen/breiten Signalen) |
|
| Alterung der Phase/Phase beschädigt durch harsche Bedingungen |
|
| Kontamination der Säule |
|
| Erhöhte Flussrate |
|
| Bei nicht wasserstabilen Phasen in hochwässrigen Eluenten: Schlechte Hydratisierung der Phase |
|
Retentionszeit ist verlängert
| Mögliche Ursachen | Lösung |
| Veränderung der Eluentenzusammensetzung |
|
| Reduzierte Flussrate |
|
Verschiebung der Retentionszeit in eine Richtung
| Mögliche Ursachen | Lösung |
| Leckage der Pumpendichtung |
|
| Überladung der Säule |
|
| Unzureichende Äquilibrierung |
|
Retentionszeit schwankt / willkürliche Verschiebung
| Mögliche Ursachen | Lösung |
| Schwankende Temperatur |
|
| Unzureichende Lösungsmittelvermischung |
|
| Falsche Pufferkonzentration oder pH-Wert |
|
| Unzureichende Äquilibrierung |
|
| Von einem System zum anderen: Unterschied in der Verweilzeit |
|
| Leckagen |
|
| Ventil an Pumpe defekt |
|
| Luft in der Pumpe, Luft in der mobilen Phase |
|
Verlust der Auflösung
| Mögliche Ursachen | Lösung |
| Kontaminierte mobile Phase |
|
| Blockierte Vorsäule |
|
| Alterung der Phase |
|
Spikes
| Mögliche Ursachen | Lösung |
| Luftblasen in mobiler Phase |
|
| Luft in der Säule |
|
Drift zu höherem Signal
| Mögliche Ursachen | Lösung |
| Anreicherung und Elution von Verunreinigungen |
|
| Viskosität der mobilen Phase zu hoch |
|
| Bei Gradienten: zunehmende Komponente B zeigt starke UV-Absorption |
|
Drift zu geringerem Signal
| Mögliche Ursachen | Lösung |
| Bei Gradienten: abnehmende Komponente A zeigt starke UV-Absorption |
|
Rauschen
| Mögliche Ursachen | Lösung |
| Wellenartig: Temperaturschwankungen im Raum |
|
| Dauerrauschen: Detektorlampe defekt oder Detektorzelle verschmutzt |
|
| Regelmäßiges Rauschen: Pumpe |
|
| Willkürliches Rauschen: Verunreinigungen |
|
| Spikes: Luft im Detektor, Eluenten oder der Pumpe |
|
Negativpeak
| Mögliche Ursachen | Lösung |
| RI-Detektor: Brechungsindex der Analyten ist geringer als von mobiler Phase |
|
| UV-Detektor: Absorption der mobilen Phase ist höher als die vom Analyten |
|
Peakflächen sind reduziert
| Mögliche Ursachen | Lösung |
| Probenverlust durch Leckage am Injektor, an Kapillaren oder Verschraubungen |
|
| Verringerte Messintensität des Detektors durch kontaminierte oder beschädigte Flusszelle/gealterte UV-Lampe |
|
| Zu geringes Injektionsvolumen |
|
| Eluent zeigt zu hohe Absorption |
|
| Verschmutzungen am Detektor |
|
Geisterpeaks
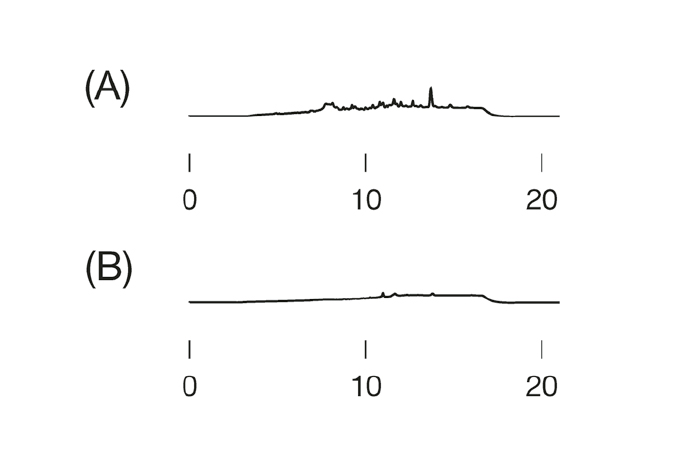
Säulen-Regenerierung
Ablagerungen auf der stationären Phase sind ein häufiger Grund für eine Beeinträchtigung der Säulenperformance. Dies äußert sich vor allem durch
• einen erhöhten Rückdruck,
• verschobene Retentionszeiten oder
• deformierte Peaks.
Um Ablagerungen zu verhindern, werden der Einsatz von Vorsäulen und eine entsprechende Probenvorbereitung wie beispielsweise Filtration empfohlen.
Sowohl zur Vorbeugung als auch dann, wenn es bereits zu einer Adsorption von Material auf der stationären Phase gekommen ist, sind Spülschritte eine effektive Maßnahme zur Regeneration der Säule. Das Spülen sollte immer gegen die Flussrichtung durchgeführt werden, da sich die Verunreinigung meist noch am Säuleneingang befindet und so leichter herausgespült werden kann. Als ausreichende Spülmenge sind mindestens 20 Säulenvolumen mit einem geeigneten Lösungsmittel empfehlenswert.
Bei der Auswahl der Lösungsmittel sollten die Eigenschaften der Verunreinigung sowie die Beständigkeit der Säule beachtet werden. In folgender Übersicht sind geeignete Lösungsmittel in Abhängigkeit von verschiedenartigen Verunreinigungen aufgeführt. Die Effizienz der Reinigung kann durch erhöhte Temperatur gesteigert werden.
Je nach eingesetztem Lösungsmittel und unter Berücksichtigung der Beständigkeit der stationären Phase sind 40 °C – 90 °C hierfür einsetzbar. Weitere Empfehlungen können auch den Anweisungen der Säulenhersteller entnommen werden, zum Beispiel den Care and Use Instructions.
Verunreinigung durch verschiedene Substanzen
|
Verunreinigung
|
Salze
|
Non-polar substances
|
Polare Substanzen
|
Proteine
|
||||
| Herangehensweise |
|
|
|
|
||||
Präventive Maßnahmen
Generell gilt:
• Regelmäßig System, Säule, Injektor spülen
• Verschleißteile wie Filter, Fritten, Dichtungen regelmäßig wechseln
um Kontamination zu vermeiden.
Zudem kann das Führen eines Säulenlogbuches die Fehlersuche stark erleichtern. Wir empfehlen neben der Identität der verwendeten Säule, Anzahl der Injektionen und dem resultierenden Rückdruck auch die verwendeten Analyten und Methoden aufzuführen und regelmäßig durch eine Testmethode die Qualität der Trennsäule an den erhaltenen Ergebnissen zu bewerten.
Es sollten auch Notizen aufgeführt werden, die den Wechsel der Eluenten, Fehler im System oder andere Auffälligkeiten hervorheben. So kann nicht nur eine möglicherweise Tage oder Wochen andauernde Fehlersuche in wenigen Minuten abgeschlossen sein, sondern auch der Produktivitätsgewinn nach einer Optimierung Ihrer Methodenparameter verfolgt werden.
Die Verwendung einer Verwendung einer Vorsäule schützt die Hauptsäule vor Kontamination, indem diese schon auf der Vorsäule adsorbiert werden.
Dadurch wird vermieden:
• Vorzeitige Alterung der Trennsäule
• Retentionszeitverschiebung
• Peakdeformation etc.
• Totvolumen in der stationären Phase durch Druckpulse
Vorsäulen sollten regelmäßig gewechselt werden, bspw. wenn der Druck steigt.
Mit einer allgemeinen Testmethode können AufSolution, Peaksymmetrie und Rückdruck aufgezeichnet und der Status von Säule und Vorsäule kontrolliert werden.

Totvolumina führen vor allem in UHPLC-Anwendungen zu Bandenverbreiterung. Um breite Peaks zu vermeiden, können totvolumenfreie Universalverbinder wie der MarvelXACT verwendet werden. Außerdem sollten alle Kapillaren im System so kurz wie möglich gehalten werden.
HPLC/MarvelXACT-Fitting-System.jpg)
Das Probenlösungsmittel kann einen starken Einfluss auf die Trennung haben, deshalb sollte Folgendes beachtet werden:
• Schwache Lösungsmittel für die Probe verwenden ➔ ansonsten Fronting früher Peaks
• Starke pH-Unterschiede zwischen Probe und Eluent vermeiden ➔ ansonsten Peak Splitting

Es ist gut zu erkennen, dass ohne ausreichende Äquilibration die Retentionszeiten deutlich nach vorne verschoben sind. Äquilibriert man mit 5 Säulenvolumen (SV) oder 10 SV, sind die Ergebnisse dieser Applikation reproduzierbar.
Wie kann die optimale Einspülzeit abgeschätzt werden?
➔ Mit Hilfe des Säulenvolumens!
Das geometrische Säulenvolumen ist ein sehr hilfreiches Mittel zur Abschätzung des notwendigen Lösungsmittel-Volumens für Spül- und Äquilibrationsschritte in der HPLC – Eine wichtige Voraussetzung für reproduzierbare und valide Ergebnisse.
Berechnung des Säulenvolumens
Säule: YMC-Triart C18
Säulendimension: 250 x 4.6 mm ID
Säulenvolumen [mL] = 25 cm × (0.23 cm)2 × 3.14
= 4.2 cm3
Was ist sonst zu beachten?
Dwell Volume
• Volumen des Systems bevor das Lösungsmittel die Säule erreicht
• Systemabhängig (Fragen Sie Ihren Hersteller)
Volumen der stationären Phase
• Korrekturfaktor (0,6–0,8) einrechnen
• Abhängig von stationärer Phase und Packdichte

Overview of geometrical column volumes [mL] for selected column dimensions

Technische Dokumente
| Titel | Dokumententyp | Sprache | Download |
 YMC (U)HPLC Troubleshooting Guide YMC (U)HPLC Troubleshooting Guide |
Brochure | EN | |
| YMC (U)HPLC Troubleshooting Handbuch |
Brochure | DE | |
 (U)HPLC Method Parameter Quick Reference (U)HPLC Method Parameter Quick Reference |
Expert Tip | EN | |
| (U)HPLC Methodenparameter Kurzübersicht |
Expert Tip | DE | |
| A hidden cause for peak tailing of small acidic compounds |
Expert Tip | EN | |
| Easy solution for distorted peaks in HPLC methods of the European Pharmacopoeia (Ph. Eur.) |
Expert Tip | EN | |
| Eine versteckte Ursache für Peak Tailing in der (U)HPLC-Analytik saurer Verbindungen |
Expert Tip | DE | |
| Einfache Lösung für gestörte Peakformen in HPLC Methoden der Europäischen Pharmakopöe (Ph. Eur.) |
Expert Tip | DE | |
| Equilibration – As much as necessary, as little as possible |
Expert Tip | EN | |
| Expert tip: Column regeneration |
Expert Tip | EN | |
| Expertentipp: Säulenregeneration |
Expert Tip | DE | |
| Guideline to buffer selection for RP methods |
Expert Tip | EN | |
| How to improve the analysis of hydrophilic compounds |
Expert Tip | EN | |
| How to improve the analysis of hydrophobic compounds |
Expert Tip | EN | |
| Is your sample soluble in your mobile phase? |
Expert Tip | EN | |
| Issues with basic analytes |
Expert Tip | EN | |
| Ist Ihre Probe in Ihrer mobilen Phase löslich? |
Expert Tip | DE | |
| Minimising carryover in oligonucleotide analysis by AEX |
Expert Tip | EN | |
| Äquilibration – So viel wie nötig, so wenig wie möglich |
Expert Tip | DE | |
 Stability of mobile phase in oligonucleotide analysis with LC-MS Stability of mobile phase in oligonucleotide analysis with LC-MS |
Experti Tip | EN | |
 The Principles of BioLC & YMC BioLC Phase Overview The Principles of BioLC & YMC BioLC Phase Overview |
Poster | EN | |
| The Principles of Liquid Chromatography |
Poster | EN | |
| Overview on YMC-Hardware Types in Combination with YMC-Phases |
Product Information | EN | |
 Allowable Adjustments to HPLC Methods in the European Pharmacopoeia (Ph.Eur.) Allowable Adjustments to HPLC Methods in the European Pharmacopoeia (Ph.Eur.) |
Technical Note | EN | |
| Erlaubte Änderungen in HPLC Methoden aus der Europäischen Pharmakopöe (Ph.Eur.) |
Technical Note | DE | |
| UHPLC systems compatible with 1 mm ID columns |
Technical Note | EN |



